Maschinelles Lernen erfasst atomare Bewegungen
Verfahren sagt Phasenübergangstemperaturen und Gitterkonstanten mit hoher Präzision vorher.
Auf atomarer Ebene können Materialien eine reiche Palette an dynamischem Verhalten zeigen, das sich direkt auf ihre physikalischen Eigenschaften auswirkt. Seit vielen Jahren versuchen Wissenschaftler diese Dynamik in komplexen Materialien bei verschiedenen Temperaturen zu beschreiben. Forscher der Uni Wien haben jetzt eine neue „On-the-fly“-Maschinenlernmethode entwickelt, die solche Berechnungen durch direkte Einbindung in das weit verbreitete Vienna Ab-initio Simulationspaket VASP ermöglicht. Die Vielseitigkeit der selbstlernenden Methode belegen neue Erkenntnisse über die Phasenübergänge von Hybrid-Perowskiten, die wegen ihres Potenzials als neuartige Solarzellen-Materialien zu dienen von großem wissenschaftlichem Interesse sind.
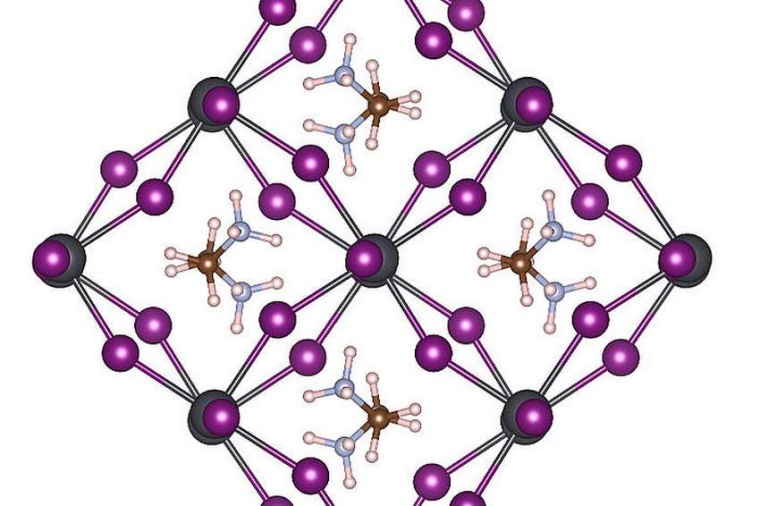
Bei Raumtemperatur befinden sich die Atome aller Materialien in ständiger Bewegung. Die physikalischen Eigenschaften von Materialien stehen in direktem Zusammenhang mit der Anordnung ihrer Atome. Je nach Temperatur oder Druck können sich diese Anordnungen und die Materialeigenschaften ändern. So ist ein Diamant transparent und hart, weil die Kohlenstoffatome im Diamantkristall periodisch angeordnet sind. Aus den gleichen Atomen entsteht bei anderer Anordnung schwarzer, spröder Graphit. Für einfache Materialien ist es möglich, die Position ihrer Atome bei verschiedenen Temperaturen mit quantenmechanischen Molekulardynamik-Simulationen genau zu bestimmen. Solche Berechnungen sind jedoch rechenintensiv und beschränken die praktische Anwendung auf ein paar hundert Atome und eine begrenzte Simulationszeit.
Die Forscher der Uni Wien haben nun einen neuen Ansatz entwickelt, der diese Einschränkungen überwindet und Simulationen komplexer Materialien für zukünftige Energieanwendungen ermöglicht. Das wird durch die Entwicklung eines effizienten und robusten datengesteuerten selbstlernenden Algorithmus erreicht und vor allem durch die direkte Integration dieses Algorithmus in das Vienna Ab-initio Simulation Package VASP. Im neuen Ansatz kann die Maschine ganz allein die wesentlichen Bestandteile für eine einfachere Beschreibung der wechselwirkenden Atome noch während der MD-Simulationen bereitstellen. Bereits nach der Berechnung von einigen hundert bis tausend Zeitschritten ist die Maschine genau genug, um eine Vorhersage der Positionen der Atome im nachfolgenden Zeitschritt zu machen.
Die Maschine ist auch in der Lage, eine Schätzung ihrer Genauigkeit für die nachfolgenden Schritte vorzunehmen. Wenn der Fehler zu hoch ist, schaltet die Maschine auf die genauen, aber rechenintensiven MD-Berechnungen um. Je mehr Simulationszeit vergeht, desto mehr lernt die Maschine und desto genauer wird sie. Auf diese Weise sind immer weniger MD-Berechnungen erforderlich, was schließlich dazu führt, dass alle Zeitschritte von der Maschine ausgeführt werden. Die Selbstlernfähigkeit bei laufendem Betrieb reduziert den Bedarf an menschlichem Eingreifen, der bei bestehenden Methoden des Maschinenlernens üblicherweise erforderlich ist.
Um die Leistungsfähigkeit der neuen Methode unter Beweis zu stellen, haben die Forscher damit Übergänge zwischen den verschiedenen Atomstrukturen des MAPbI3-Perowskits nach Änderung der Temperatur untersucht. Dieses Material ist wegen seines Potenzials als neues, billiges Solarzellen-Material eingesetzt zu werden sehr beliebt. Es besteht aus organischen Molekülen, die sich schnell drehen können und durch ein Gitter aus Blei- und Iodidatomen getrennt sind. Je nach Temperatur entstehen drei verschiedene Kristallphasen. Die atomaren Mechanismen in der Nähe der Übergangstemperatur sind sehr schwer experimentell zu bestimmen, und herkömmliche MD-Simulationen würden selbst auf einem modernen Supercomputer jahrelange Rechenzeit erfordern. Mit der neuen „On-the-Fly“-Methode kann die Maschine nach dem Lernen die Phasenübergangstemperaturen und Gitterkonstanten dieses Materials mit beispielloser Präzision vorhersagen. Die neu entwickelte Methode ist allgemein und auf viele andere zukünftige materialwissenschaftliche Probleme anwendbar und wird in der kommenden Version von VASP für Forscher weltweit verfügbar sein.
U. Wien
Weitere Infos
- Originalveröffentlichung
R. Jinnouchi et al.: Phase Transitions of Hybrid Perovskites Simulated by Machine-Learning Force Fields Trained on the Fly with Bayesian Inference, Phys. Rev. Lett. 122, 225701 (2019); DOI: 10.1103/PhysRevLett.122.225701 - Computergestützte Materialphysik, Fklt. Für Physik, Universität Wien, Österreich